A síndrome de Alstrom (AS), descrita por este médico em 1959, caracteriza-se principalmente pela associação de défice visual e auditivo progressivo, obesidade e diabetes. Alterações cardíacas, renais, hepáticas, pulmonares, lipídicas também são frequentes. A maioria não tem défice cognitivo, podendo ocorrer atraso na motricidade e linguagem e dificuldades de aprendizagem. Transmite-se de forma autossómica recessiva e é causada por alteração no gene ALMS1, mas em cerca de 20% a causa ainda é desconhecida. Estima-se uma prevalência de 1/1.000.000, mas é possível que esteja sub-diagnosticada.
Clínica
É uma doença que atinge vários órgãos, cujas manifestações surgem ao longo da vida:
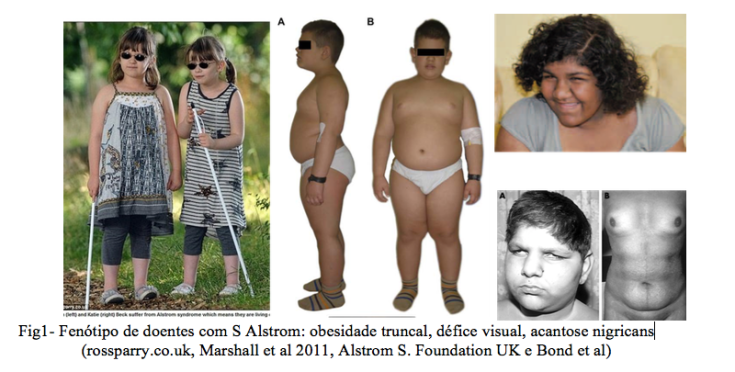
Distrofia da retina de tipo cone e bastonete (100%) com consequente nistagmo (movimentos oculares involuntários) e fotofobia (hipersensibilidade à luz) de início no primeiro ano de vida e com défice visual progressivo; muitos doentes perdem a percepção de luz na segunda década, mas alguns conseguem ler grandes letras até à terceira década.
Obesidade (98%) inicia-se na infância e é essencialmente do tronco. Ao nascer o peso é habitualmente normal
Surdez neurosensorial progressiva (>80%), moderada a severa, tem início na 1ª década.
Insulinoresistência (>90%), traduzindo-se por hiperinsulinemia, intolerância á glicose e diabetes tipo 2 (70%) com início na infância ou adolescência. O aparecimento de lesões de hiperpigmentação cutânea (acantose nigricans) é indicador de insulinorresistência.
Cardiomiopatia dilatada ou restritiva (>60%) pode ocorrer em qualquer idade.
Doença pulmonar: infecções recorrentes são frequentes desde a infância e mais tarde pode existir insuficiência respiratória por fibrose pulmonar
Alterações uronefrológicas (50%) incluem disfunção vesico-uretral com retenção ou incontinência e alterações do interstício renal que podem conduzir a insuficiência renal
Doença hepática traduz se por enzimas hepáticos frequentemente elevados na infância, eventual hepatomegalia e esteatose hepática e possível evolução para cirrose na segunda-terceira década.
Hiperlipidemia é comum, sobretudo por aumento dos triglicéridos
Hipogonadismo ocorre em 80% dos homens por défice de hormonas sexuais (de causa central e por fibrose testicular), originando atraso pubertário, ginecomastia e provável infertilidade. Na mulher existe baixo nível de gonadotrofinas e excesso de androgénios e é frequente ovários poliquisticos, puberdade precoce, irregularidades menstruais ou amenorreia. A fertilidade deverá estar diminuída, mas há algumas mulheres com descendência.
Hipotiroidismo ocorre em 20% dos doentes
Perturbação do neurodesenvolvimento: cerca de 20% têm atraso na aquisição da motricidade grosseira e fina e também linguagem, e 30% dificuldade de aprendizagem, mas é raro existir défice cognitivo. Ouras alterações (perturbação do espectro do autismo, epilepsia…) são ocasionalmente referidas.
Baixa estatura: a estatura em adulto está geralmente abaixo do percentil 5
Escoliose, cifose e pés planos também são referidos
Geralmente não existem dismorfias importantes, sendo o fenótipo externo marcado pela obesidade, as perturbações da visão e as alterações cutâneas de acantose nigricans (Fig1).
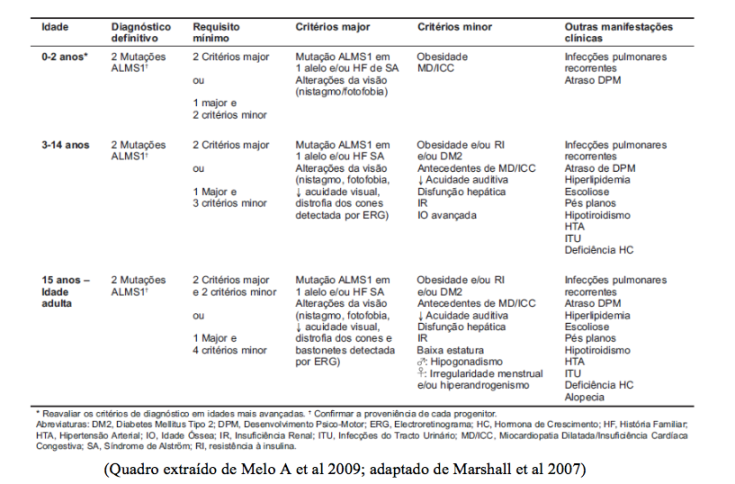
O diagnóstico clínico baseia-se nas características principais, acima mencionadas, e que surgem ao longo da infância, adolescência e idade adulta, tendo sido definidos critérios de diagnóstico de acordo com a idade:
Etiologia
A S. Alstrom é causada por mutação ou deleção no gene ALMS1 de transmissão autossómica recessiva. Em cerca de 20% dos doentes com critérios clínicos ainda não é possível encontrar a causa. A proteína codificada por este gene tem expressão nos cílios de células dos vários órgãos atingidos, pelo que a AS pertence ao grupo das ciliopatias
Diagnóstico
Existindo uma suspeita com base nos critérios clínicos, deverá proceder-se ao estudo do gene ALMS1, sendo esperado encontrar alteração nas duas cópias do gene. Testes negativos (sequenciação e pesquisa de deleções) não invalidam o diagnóstico, uma vez que em cerca de 20% dos doentes não se encontram alterações neste gene. No entanto, será importante rever os doentes no sentido de eventual diagnóstico diferencial com situações com clínica semelhante como a Síndrome de Bardet-Biedl.
Aconselhamento Genético
Sendo uma situação de transmissão autossómica recessiva, os pais (saudáveis) serão portadores de mutação em apenas 1das 2 cópias do gene (heterozigotos), pelo que a probabilidade de terem outros filhos com a mesma doença é de 25%, sendo possível diagnóstico pré-natal ou pré-implantatório desde que as mutações tenham sido identificadas no doente. Os doentes com AS têm fertilidade reduzida (sobretudo no sexo masculino) mas se tiverem filhos, estes serão possivelmente saudáveis (heterozigotos), excepto se o parceiro for doente ou familiar de doente, caso em que terão alta probabilidade de ter filhos com a mesma situação.
Medidas de Vigilância e Terapêutica
Estes doentes deverão ser seguidos por equipa multidisciplinar que inclua as especialidades de Pediatria Geral, Neurodesenvolvimento, Clínica Geral, Oftalmologia, Cardiologia, Endocrinologia, ORL, Nefro-urologia Gastroenterologia, Pneumologia, de forma a tentar prevenir, vigiar, tratar ou minorar os problemas referidos e que poderão surgir ao longo da vida. É essencial prestar atenção precocemente à motricidade, linguagem, e aos défices sensoriais (visão e audição) planeando formas alternativas de ensino e de apoio para as actividades diárias (exemplo: Braille, cães guias, próteses auditivas…)
Referências
-Alstrom CH et al: Retinal degeneration combined with obesity, diabetes mellitus and neurogenous deafness: a specific syndrome (not hitherto described) distinct from the Laurence –Moon–Bardet –Biedl syndrome: a clinical, endocrinological and genetic examination based on a large pedigree. Acta Psychiatr Neurol Scand Suppl 1959;129: 1– 35.
– Bond J et al.The importance of seeking ALMS1 mutations in infants with dilated cardiomyopathy. J Med Genet. 2005; 42:e10
– Collin GB. et al. Mutations in ALMS1 cause obesity, type 2 diabetes and neuro sensory degeneration in Alstrom syndrome. Nat Genet 2002;31:74-8.114
-Hearn T. et al. Mutation of ALMS1, a large gene with a tandem repeat encoding 47 amino acids, causes Alstrom syndrome. Nat Genet 2002;31:79-83.
-Makaryus A. et al. Cardiac manifestations of Alstrom syndrome: echocardiographic findings. J Am Soc Echocardiogr 2007;20:1359-63.
-Marshall JD. et al. New Alstrom syndrome phenotypes based on the evaluation of 182 cases. Arch Intern Med 2005;165:675-83.
-Marshall JD et al. Alstrom syndrome. Eur J Hum Genet 2007a;15:1193–202.
-Marshall JD. et al. Spectrum of ALMS1 variants and evaluation of genotype-phenotype correlations in Alstrom syndrome. Hum Mutat 2007b;28:1114-23.
-Marshall JD. et al. Alstrom Syndrome: Genetics and Clinical Overview. Curr Genom. 2011; 12(3):225–235.
– Marshall JD. et al. Alström Syndrome: Mutation spectrum of ALMS1. Hum Mutat. 2015 July; 36(7): 660–668
– Melo A. et al. Síndrome de Alström. Acta Pediatr Port 2009:40(3):111-5
-Ozantürk A et al. The phenotypic and molecular genetic spectrum of Alström Syndrome in 44 Turkish kindreds and a literature review of Alström Syndrome in Turkey. J Hum Genet. 2015 January ; 60(1): 1-9.
-Paisey RB et al. Hypertriglyceridaemia in Alström Syndrome: causes and associations in 37 cases. Clin Endocrinol.2004 Feb;60(2):228-31.
-Richardson D et al Renal diagnosis without renal biopsy. Nephritis and sensorineural deafness Nephrol Dial Transplant 2001;16:1291-4.
-Satman I.et al. Evaluation of insulin resistant diabetes mellitus in Alstrom syndrome: a long-term prospective follow-up of three siblings. Diabetes Res Clin Pract 2002;56:189-96.
– Tisha J et al. Alstrom syndrome (OMIM 203800): a case report and literature review. Orphanet J Rare Dis 2007;2:49.

Deixe um comentário